Transcription:
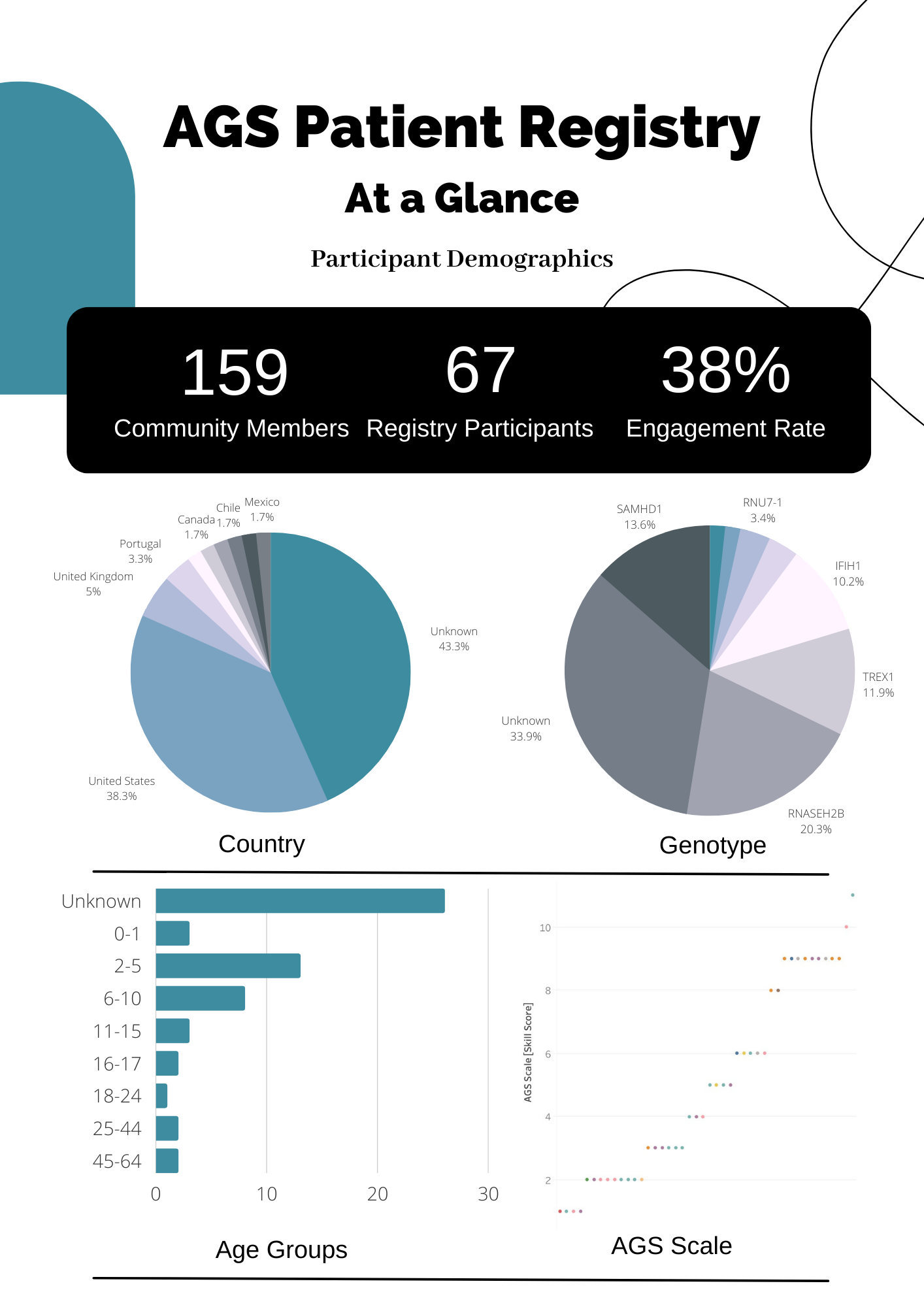
Hi, everyone. I wanted to introduce myself and take a moment to give you some perspective on the AGS patient registry managed by the Aicardi Goutieres Syndrome Advocacy Association (the AGSAA). My name is Patrick, and I've taken on the responsibility of research director at the AGSAA. I'm a father of three children, the youngest of which has been impacted significantly by AGS. From the very beginning of this journey, I've dedicated myself to understanding this disease and working towards solutions to improve my daughter's care. I've recently taken the lead in communications and collaborations with scientists and clinicians working on AGS. And, one of the most important and compelling things we can offer potential collaborators is coordinated access to patients, data, samples, et cetera. This registry represents and reflects our rare disease community's level of organization. A scientist looking to research a disorder or a drug company evaluating the cost of pursuing treatments for a disease will consider this in their decisions. Over the next year I've volunteered to serve as co-chair for the Global Leukodystrophy Initiative's Coalition of Patient Advocacy Groups. In my involvement with GLIA, the National Institute of Health in the U.S. , and the Rare Disease Clinical Research Network, I'll be working to help other rare disease groups prepare themselves for clinical trials by establishing registries, just like ours.
The establishment of a patient registry is the number one recommendation made by these institutions. It's the most important thing advocacy groups can do to prepare for the development of future clinical trials. Since we launched the registry, we've tried to focus surveys on data and topics that would allow us to provide useful information to families quickly and directly. By doing so, we've hoped to build a group of engaged and active participants, a necessary precursor to attracting scientists for more in depth research.
Still, I believe there's compelling work and novel insights that we can publish directly. For example, our triggers and flare survey, which is open now, will allow us to publish a full and detailed characterization of AGS disease flares. And, this is something that isn't currently written about nor well understood by families or their doctors. I'd also like to comment on data and access and visibility. None of us. And I mean it, none of us have the ability to view your name, email address, or any other personally identifiable information. Luna, the software service we use, ensures that our registry remains anonymous to us and to every collaborator that we may invite to analyze the data. All of this puts our registry above board, making it compelling to a researcher that would be able to include registry information directly in their publications.
Our registry is official in that sense, it's considered reliable; and with it we're going to advance understanding and care for AGS. So we invite all families around the world to participate. Although we acknowledge that there will be some friction and shortcomings at times, we are still dedicated to making this an inclusive and comprehensive tool for our community.
I encourage and implore you to participate.